摘要:以Mo、Y、Al和C元素粉为原料, 用放电等离子烧结技术(SPS)在1550 ℃合成了新颖的(Mo2/3Y1/3)2AlC MAX相, 并用较温和的化学刻蚀方法剥离得到相应手风琴状形貌的MXene。采用X射线衍射(XRD)、扫描电子显微镜(SEM)、透射电子显微镜(TEM)和能谱分析(EDS)手段对材料的化学组成、微观结构等进行了表征, 确定最终产物为表面带有官能团的Mo1.33CT2 MXene。同时利用第一性原理密度泛函理论计算方法研究了新颖(Mo2/3Y1/3)2AlC MAX相以及对应的Mo1.33CT2 MXene的电子结构和性能, 计算结果表明两者均呈现出金属特性, 有望应用于储能、生物传感器和电催化等方面。
关 键 词: MAX; MXene; 第一性原理; 电子性能
自2004年二维石墨烯(2D graphene)被发现以来, 其独特的二维结构和物化性能备受人们关注, 随后一系列二维材料相继被报道出来, 如六方氮化硼(BN), 过渡金属二硫化物(TMDs), 金属氧化物和金属氢氧化物等[1-4]。2011年Naguib等[5]在室温下用HF化学刻蚀的方法选择性的刻蚀掉Ti3AlC2 MAX相(其中M为前过渡金属, A主要为IIIA或者IVA族元素, X是C和/或N, n=1、2或者3)的Al原子层, 得到了表面带有O/F/OH官能团的Ti3C2Tx 2D材料。因其与石墨烯的结构相似性, 该类材料被命名为MXene, 化学表达式为Mn+1Xn。目前已报道的MXene材料有20余种, 如Ti3C2、Ti2C、(Ti0.5Nb0.5)2C、(V0.5Cr0.5)3C2、Ti3CN、Ta4C3、Nb2C、V2C、Mo2C和Nb4C3等[6-8]。新型2D MXene作为二维材料具有一些独特的性质, 例如: 独特的物理化学性质, 大的比表面积, 独特的层状结构, 特殊的表面官能团等[9-11]。这些特性使其在很多方面有着潜在的应用, 近年来吸引了大量的学者对其进行实验和理论的研究[12-15]。
目前选择性刻蚀MAX相的A层元素得到MXene方法已经有很多种, 并且比较成熟。Alhabeb 等[16]用不同浓度的HF对Ti3AlC2相进行剥离, 发现HF的浓度越高剥离得越彻底, 得到的MXene二维材料越薄; 温和的刻蚀剂LiF+HCl在动态环境中也可以得到MXene, 其产物与低浓度的HF得到的形貌相似。Xuan等[17]用有机碱四甲基氢氧化铵(TMAOH)作为刻蚀剂刻蚀掉Ti3AlC2的Al原子层, 得到了Ti3C2 MXene。Urbankowski 等[18]在2016年首次在高温熔融氟盐氩气氛围中550 ℃选择性刻蚀掉Ti4AlN3的Al层, 成功得到了Ti4N3 MXene。
到目前为止, 报道的有关含Mo的三元MAX相有Mo2GaC和Mo2Ga2C。第一个含Mo的三元MAX相是Mo2GaC, 由Toth等[19]于1967年用Mo粉、C粉、液体Ga在真空石英管中850 ℃下合成。由于这种方法费时, 后来Hu 等[20]将Mo粉和C粉先混合均匀, 再与Ga混合均匀, 900 ℃氩气氛围烧结24 h得到了Mo2GaC, 合成方法较之前节省时间。继而他们又合成了M2Ga2C, 先将Mo粉和C粉按化学计量比在氩气氛围中以1000 ℃烧结12 h, 将烧结后的样品与Ga混合, 在850 ℃烧结48 h, 得到Mo2Ga2C[21]。Halim 等[22]用HF对Mo2Ga2C进行剥离, 并且成功地剥离掉Ga原子层, 得到Mo2C MXene。另外, 在合成三元MAX相时, 可以在MAX的M、A或者X位进行固溶, 来稳定原本不太稳定的MAX相, 还可以添加超过传统MAX相使用的元素, 得到一种新的固溶MAX相。Mo-Al-C三元MAX相到目前为止只有在理论计算上证明它的存在性, 其合成方法还比较困难。于是考虑合成含Mo的四元MAX 相。Anasori 等[23]合成了Mo2TiAlC2, 结构是Ti原子处于两个Mo原子之间, C原子处于Mo和Ti之间。此外, 理论计算方面对已有MAX和MXene的相关性能做了大量的研究工作, 并预测了后续有可能实验合成出的MAX和MXene材料体系, 对实验指导具有极其重要的意义[24]。Meshkian 等[25]用第一性原理的方法研究了Mo2ScAlC2的稳定性以及对应的MXene的稳定性, 证明它们都是可以稳定存在的。Tao 等[26]用第一性原理方法预测(Mo2/3Sc1/3)2AlC是稳定存在的, 合成出了这种新颖的MAX相, 且选择性地刻蚀掉了Al和Sc原子, 得到了新颖的Mo1.33C MXene。Dahlqvist 等[27]对新颖(Mo2/3Y1/3)2AlC MAX相进行了理论预测发现其声子谱没有虚频, 证明了该材料是可以稳定存在的, 并将元素粉用管式炉在1600 ℃合成出(Mo2/3Y1/3)2AlC MAX相。
目前, 关于(Mo2/3Y1/3)2AlC的块体烧结和化学剥离尚未有文献报道。本研究通过放电等离子烧结(SPS)的方法合成出新颖的(Mo2/3Y1/3)2AlC MAX相材料, 采用较温和的化学刻蚀方法得到了相应的2D Mo1.33CT2 MXene。同时利用第一性原理密度泛函理论计算方法研究了新颖(Mo2/3Y1/3)2AlC MAX相以及对应的Mo1.33CT2 MXene的电子结构和 性能。
1 实验方法
本实验将Mo, Y, Al(钼粉, 钇粉, 铝粉购买于有色金属研究院, 99.9%, 48 μm), C(青岛欧尔石墨有限公司, 99%, 74 μm)四种元素粉体按照化学计量比Mo : Y : Al : C=4 : 2 : 3 : 3混合均匀后装入石墨模具, 利用SPS在1550 ℃、, 17 MPa的氩气气氛条件下成功制备得到(Mo2/3Y1/3)2AlC块体。将块体材料用砂纸打磨移除表面杂质、破碎、研磨、过37 μm筛, 得到刻蚀所需的粉体。将0.5 g (Mo2/3Y1/3)2AlC粉体加入到LiF(1.436 g)+HCl(12 mol/L, 10 mL)的混合液中, 在35 ℃下搅拌反应100 h, 待反应结束后洗涤、离心即得2D Mo1.33CT2 MXene。采用XRD (X射线粉末衍射仪D8 ADVANCE)、配有DES的SEM (FEI Quanta FEG 250)和TEM(Tecnai F20 )对材料的物相组成、晶体结构和微观形貌进行表征。
采用第一性原理密度泛函理论(Density Functional Theory, DFT), 均通过VASP软件进行计算工作[28-29]。赝势采用投影缀加平面波(PAW)形式, 对应平面波的截断能取为 500 eV应用于Mo 4p64d55s1, Y4p64d15s2, Al3s23p1, C 2s22p2电子。交换关联泛函采用的是广义梯度 GGA-PBE 形式。MAX相和 MXene 结构优化方法采用的是共轭梯度方法。在优化过程中, 原子弛豫的收敛标准为0.01 eV/atom, 能量的收敛标准为1.0×10-5 eV/cell。为模拟二维 MXene及单层MXene结构, 消除层间相互作为的影响, 在垂直MXene平面方向施加不少于 2 nm 的真空层。MAX相的计算采用3×5×2的布里渊区K点, MXene采用6×6×1的K点阵[30-31]。
2 结果与讨论
2.1 (Mo2/3Y1/3)2AlC的合成及其电子性能分析
图1(a)~(b)分别是实验测得的(Mo2/3Y1/3)2AlC的XRD图谱与理论预测的XRD图谱。模拟的(Mo2/3Y1/3)2AlC的XRD图谱与文献[27]结果一致。通过对比发现实验数据与理论计算相吻合。由于SPS的结构设计比气氛管式炉的密封效果更好, 烧结时间短, 所以本次工作采用SPS的烧结时间为1.5 h, 比文献[27]中气氛管式炉烧结时间缩短了8.5 h。此外, 实验测得的XRD结果表明除(Mo2/3Y1/3)2AlC的峰外还有Mo2C、Mo3Al2C、Y2O3和YAlO3等杂质相。但上述杂质相对MAX相的刻蚀并无明显影响。
图2是(Mo2/3Y1/3)2AlC的晶体结构模型图, (Mo2/3Y1/3)2AlC MAX相与传统三元层状MAX相在晶体结构上有明显区别。传统MAX相的空间群是P63/mmc, 而该MAX相的空间群是c2/c, 称为面内化学有序MAX相(In-plane chemically ordered MAX phases, i-MAX), 但是它与所有的211相一样由 MX层与A层在c方向上交替堆垛而成, 也就是由(Mo2/3Y1/3)2C层与Al层交替堆垛[27]。图3是(Mo2/3Y1/3)2AlC电子态密度(DOS)和局域电子态密度(PDOS)图谱, 费米面能量设为0 eV。由图3可以看出(Mo2/3Y1/3)2AlC的总DOS跨越费米能级, 说明其呈金属属性。C 2p轨道与Mo和Y 的5s4p4d轨道在-6 eV到-3 eV之间重叠明显, 这表明M-C之间的结合形成化学键。Al 3s3p轨道与金属Mo Y5s4p4d轨道之间重叠较少, 说明M-Al之间的结合较弱。根据之前的文献报道, 该材料可通过刻蚀剂选择性地刻蚀掉MAX相材料中与M结合较弱的A原子层(Al, Ga), 从而得到相应的二维材料[17, 22], 这与理论计算的结果是一致的。
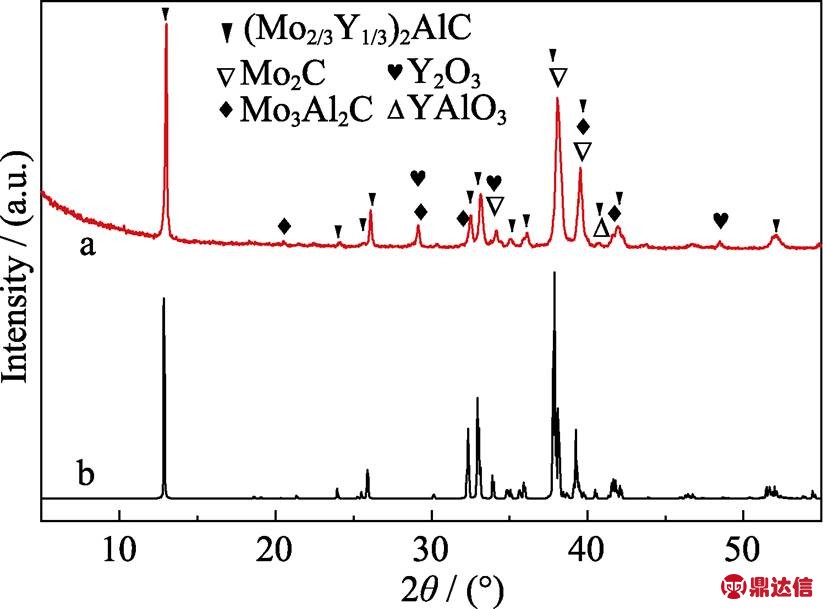
图1 (Mo2/3Y1/3)2AlC的XRD图谱(a)测试结果, (b)模拟计算结果
Fig. 1 Experimental (a) and simulated (b) XRD patterns of (Mo2/3Y1/3)2AlC
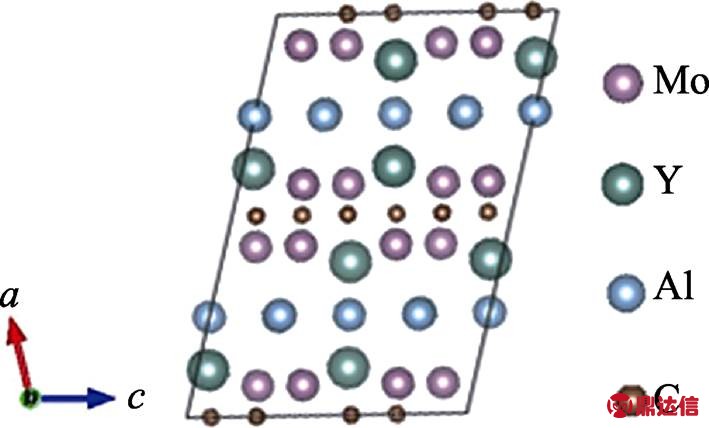
图2 (Mo2/3Y1/3)2AlC的晶体结构模型图
Fig. 2 Crystal structure of (Mo2/3Y1/3)2AlC
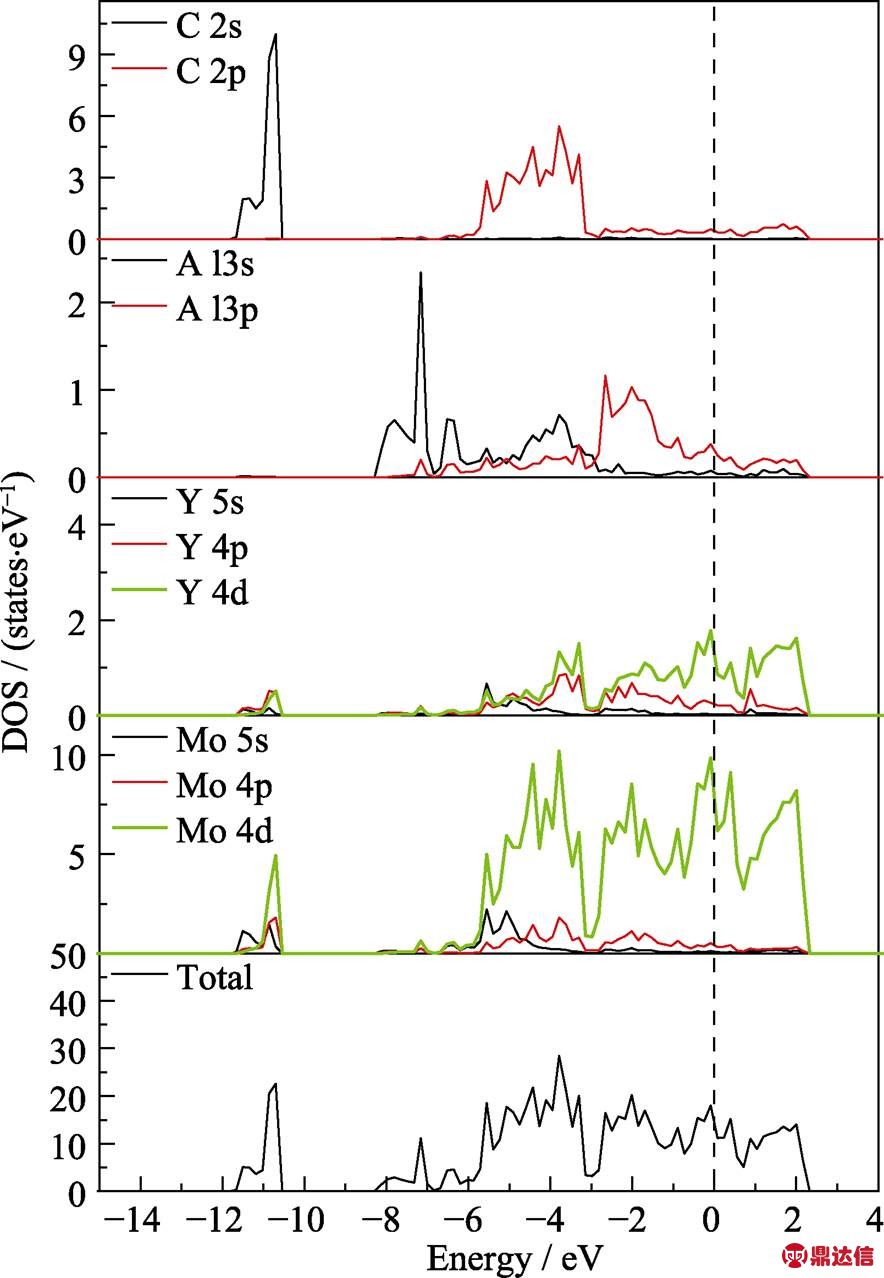
图3 (Mo2/3Y1/3)2AlC的DOS和PDOS图谱
Fig. 3 DOS and PDOS of (Mo2/3Y1/3)2AlC
2.2 (Mo2/3Y1/3)2AlC的剥离及表征
图4是(Mo2/3Y1/3)2AlC刻蚀前后的XRD图谱。图中观察到原211相特征峰(002)面在2q=13.1°, 反应后在2q=13.1°处的衍射峰基本观察不到, 在2q= 6.2°处出现了一个新的(002)面衍射峰, 与原峰相比向低角度偏移了6.9°。另外在刻蚀后的XRD图中检测到的其它衍射峰为Mo2C、Mo3Al2C和Y2O3。反应前原211相的特征峰(002)面晶面间距为0.67 nm, 反应后晶面间距1.19 nm, 晶面间距增大了0.52 nm。文献[26]中(Mo2/3Sc1/3)2AlC剥离后晶面间距从1.39 nm增至1.94 nm, 增大了0.55 nm, 晶面间距增加比较接近。可以确定得到了对应的二维材料。图5是(Mo2/3Y1/3)2AlC反应前后的SEM照片和TEM照片。图5(a)是反应前(Mo2/3Y1/3)2AlC的SEM照片, 从图中可以看出在侧面有层状痕迹, 说明该材料是典型的层状结构。图5(b)是(Mo2/3Y1/3)2AlC在LiF+HCl中进行刻蚀后的SEM照片, 可以清楚的看到手风琴状的形貌。用EDS进一步分析结果表明产物中各元素的原子比为Mo : Y : Al : C : O : F : Cl≈21.87 : 0.54 : 1.28 : 16.57 : 12.49 : 21.20 : 26.05, Y的含量只有0.54at%, Al的含量只有1.28at%, 可以认为二者基本都已经被刻蚀掉, Mo与C的原子比约为1.33 : 1.00, 与文献[26]中报道得到Mo1.33C的结果是相似的, 即MAX相中的Al被刻蚀的同时Y也被刻蚀掉了, 最终得到的产物为Mo1.33CTx (T=O/F/OH), 与XRD结果吻合。将0.5 g Mo1.33CTx与10 mL 25%四甲基氢氧化铵(TMAOH)在室温下搅拌24 h进行插层。图5(c)是经TAMOH插层处理后得到的Mo1.33CTx的TEM照片, 可观察到对电子束透明的单层或几层的薄片, 在图5(c)中所示片层可以明显看出褶皱, 这说明得到了二维材料, 与之前报道的Ti2CTxMXene相似[13]。
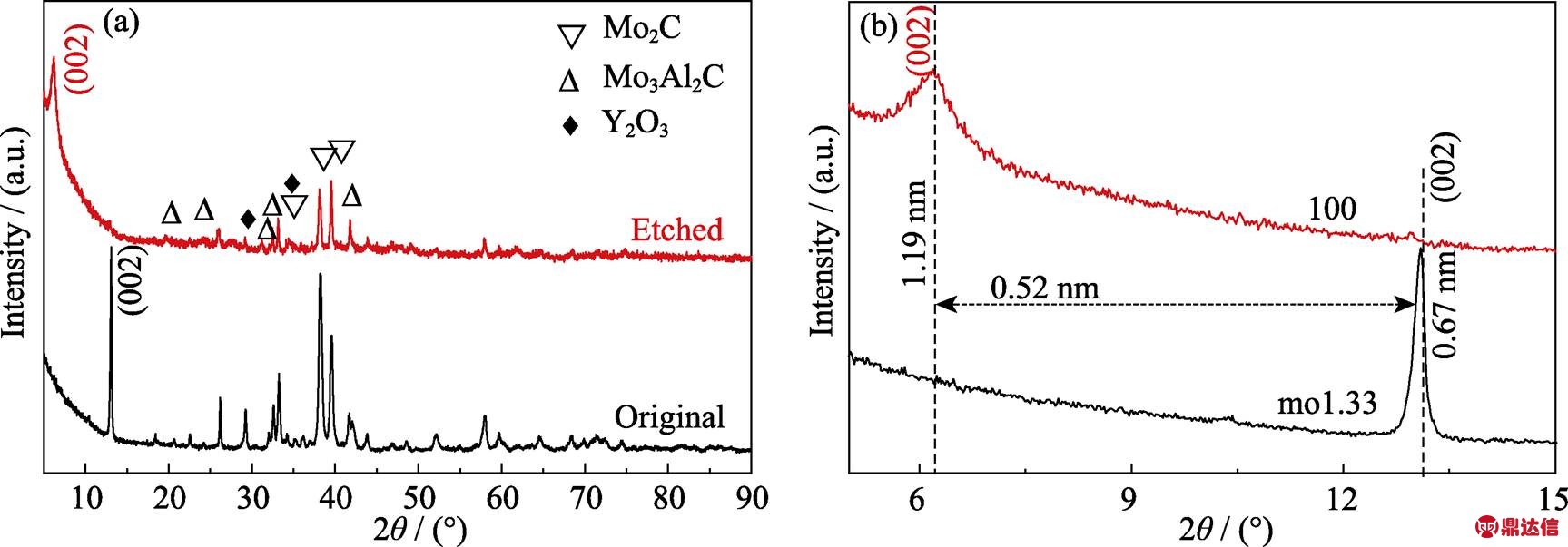
图4 (Mo2/3Y1/3)2AlC 在刻蚀前后的XRD图谱, (b)是(a)的低角度放大图谱
Fig. 4 XRD patterns of (Mo2/3Y1/3)2AlC before and after etching with (b) low-angle magnification of (a)
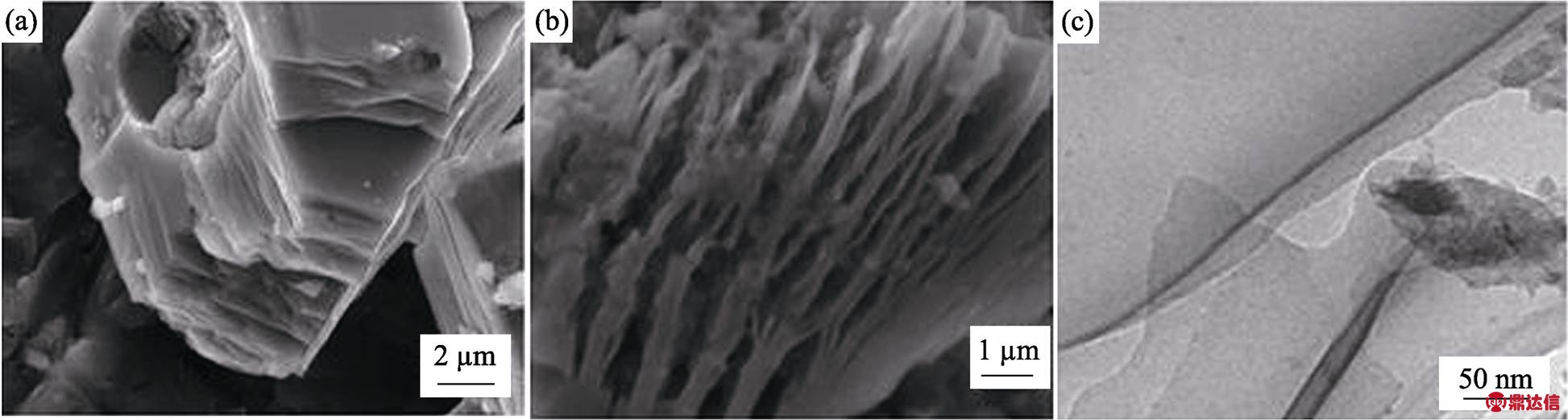
图5 (a) (Mo2/3Y1/3)2AlC和(b) Mo1.33CTx的SEM照片, (c)Mo1.33CTx的TEM照片
Fig. 5 SEM images of (a) (Mo2/3Y1/3)2AlC and (b) Mo1.33CTx, (c) TEM image of Mo1.33CTx
2.3 Mo1.33CT2的电子性能
根据文献[32]报道的结果可以确定Mo1.33C的官能团数为2。经计算发现表面官能团的位置处于图6所示位置时最稳定。图6是Mo1.33CT2的晶体结构模型图。Mo1.33C可以看作是Mo2C MXene中每隔两个Mo原子去掉一个Mo原子。在本次工作中对不同表面官能团Mo1.33C的DOS和PDOS进行了计算。图7中(a)~(c)分别是表面官能团为F, O, OH的DOS和PDOS图谱, 把0 eV设为费米面, 从图中可以看出当Mo1.33C的表面基团是F, O, OH时都为导体, 这与文献[32]中的结果一致。这可能是由于刻蚀掉Y原子后体系中空位的存在使得体系中的电子富余, 更加容易导电。它们导电的电子主要是由Mo 4d轨道贡献的, Mo的4d轨道与C的2p轨道在-6 eV到-1 eV之间重叠较多, 这说明Mo与C之间的结合较强。Mo的4d轨道与O的2p轨道在-7 eV到-1 eV之间重合较多, Mo的4d轨道与F的2p轨道在-7 eV到-3 eV之间重叠较多, 这说明Mo与O、F之间的结合较强。相比之下, OH与Mo的4d轨道的重合较少, 说明Mo与OH的结合较弱, 多种表面基团同时存在时, OH更容易被其它表面基团所替换。
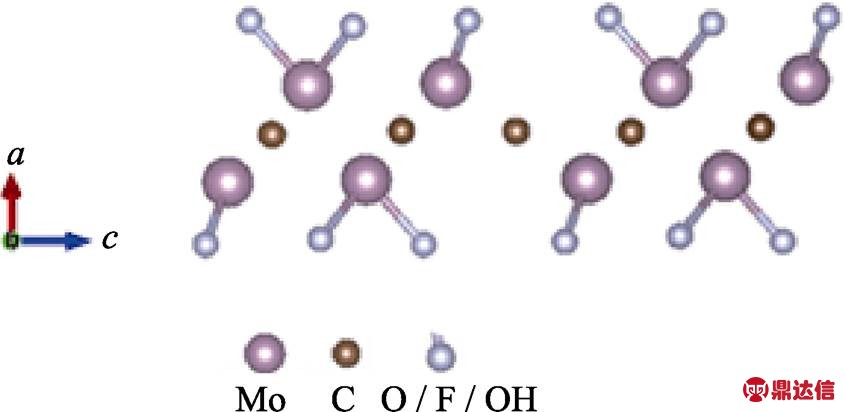
图6 Mo1.33CT2 (T=O/F/OH)的晶体结构
Fig. 6 Crystal structure of Mo1.33CT2 (T=O/F/OH)
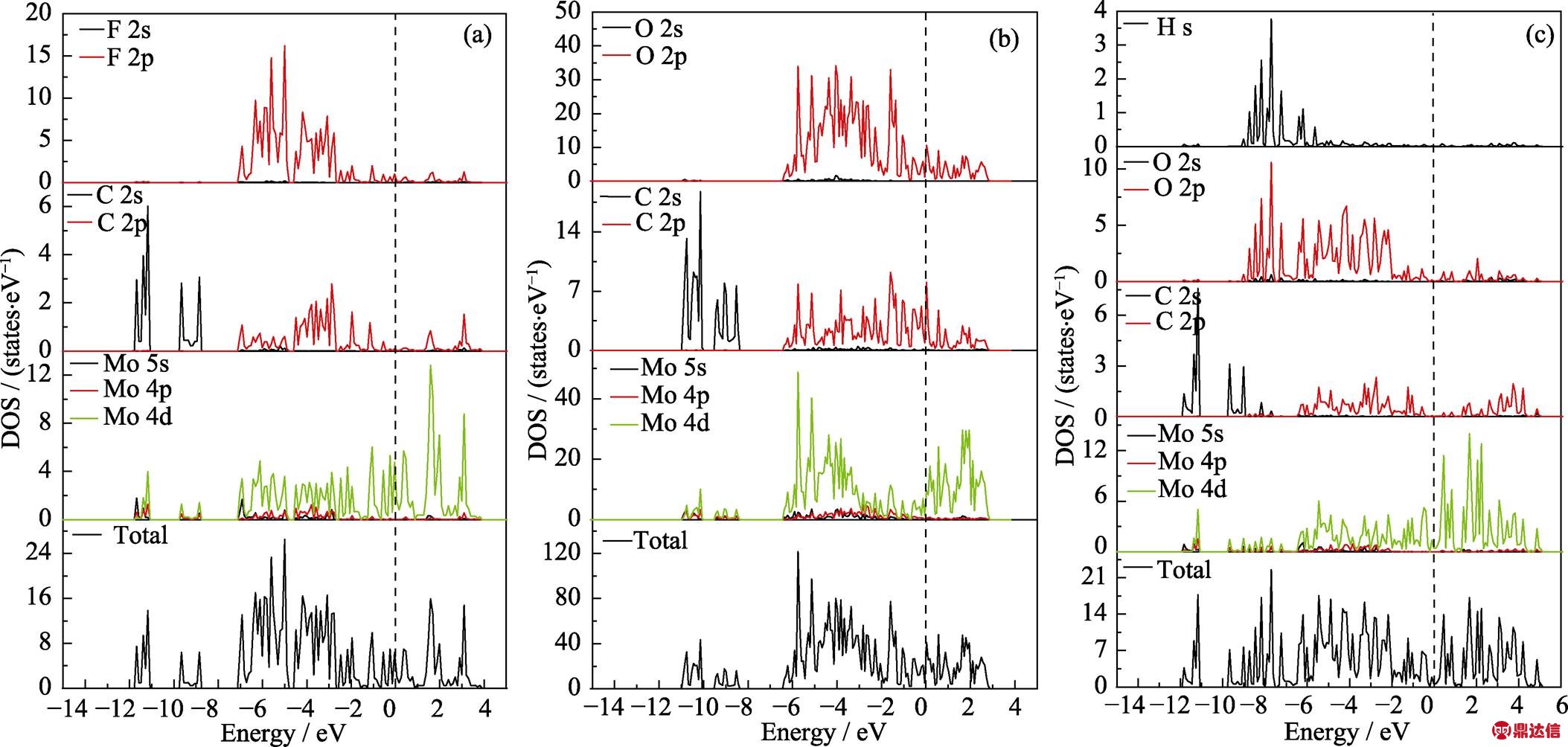
图7 Mo1.33CT2的DOS和PDOS图谱
Fig. 7 DOS and PDOS of Mo1.33CT2
(a) T=F; (b) T=O; (c) T=OH
2.4 Mo1.33CT2的弹性常数
实验结果表明得到的Mo1.33CT2 MXene具有柔性, 因此采用第一性原理方法计算了Mo1.33CT2在不同表面官能团下的弹性常数(C11)。MXene的C11值可以按照下面公式计算[15,33]。
C11=C11cell × c/dL (1)
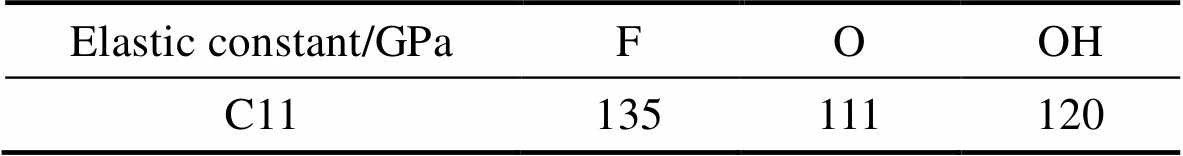
在公式(1)中C11cell是计算用晶胞的C11, c是c方向上的晶格常数, 在本次计算中为2 nm, dL是层厚, T=F、O和OH分别为0.4949、0.4730和0.6916 nm。本次计算工作中获得的Mo1.33CT2 C11的值列于表1, 从表中可以看到当T=F、O和OH时, C11分别为135、111和120 GPa。Zha等[33]计算了M2CT2 (M=Sc, Ti, V, Cr, Zr, Nb, Mo, Hf, Ta, W)的C11值, 当T=F, O, OH时分布在80~500 GPa之间, 本次工作中计算的Mo1.33CT2的C11值处于这个区间, 但是偏小, 可能是由于每隔两个Mo原子就存在一个空位, 正是由于空位的存在使得它的机械强度降低, 所以C11的值比Mo2C的小, 但是能够说明得到的Mo1.33CT2具有一定的机械强度。
表1 Mo1.33CT2的弹性常数
Table 1 Elastic constant of Mo1.33CT2
3 结论
本工作采用SPS方法在1550 ℃氩气气氛下合成了新颖的(Mo2/3Y1/3)2AlC MAX相, 通过温和的化学刻蚀方法对合成的新颖(Mo2/3Y1/3)2AlC材料进行刻蚀, 实验结果表明在Al元素被刻蚀掉的同时Y元素也被刻蚀掉。最后得到的产物为带表面官能团的新颖2D Mo1.33CT2(T=F/O/OH)。同时用第一性原理对MAX和Mo1.33CT2 MXene的电子性能进行了研究, DOS和PDOS的结果表明两者均呈金属属性。计算其弹性常数表明Mo1.33CT2具有一定的机械强度。得到的最终产物是导电二维碳化钼MXene材料Mo1.33CT2, 其独特的二维层状结构以及金属特性有望应用于储能、生物传感器和电催化等方面。